SPARK is a R package developed to identify genes with spatial expression pattern in spatially resolved transcriptomic studies.
SPARK
SPARK directly models count data generated from various spatial resolved transcriptomic techniques through generalized spatial linear models. It relies on penalized quasi-likelihood algorithm for scalable computatation and recently developed statistical formulas for hypothesis testing, providing effective control of type I errors and yielding high statistical power.
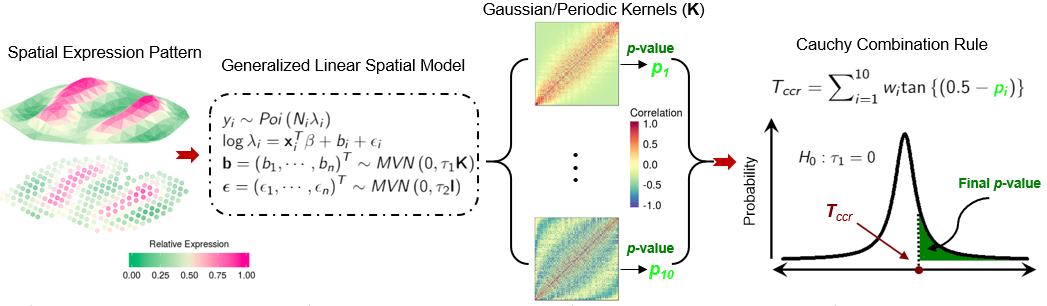
Recommend Application: Sample size smaller than 3,000, with relatively low sparsity structure. Example Analysis with SPARK:here.
SPARK-X
SPARK-X builds upon a robust covariance test framework to model a wide variety of spatial transcriptomics data collected through different technologies. It relies on algebraic innovations for scalable computatation as well as newly developed statistical formulas for hypothesis testing, producing well-calibrated p-values and yielding high statistical power. SPARK-X is highly computationally efficient and the only SE method scalable for the HDST data.
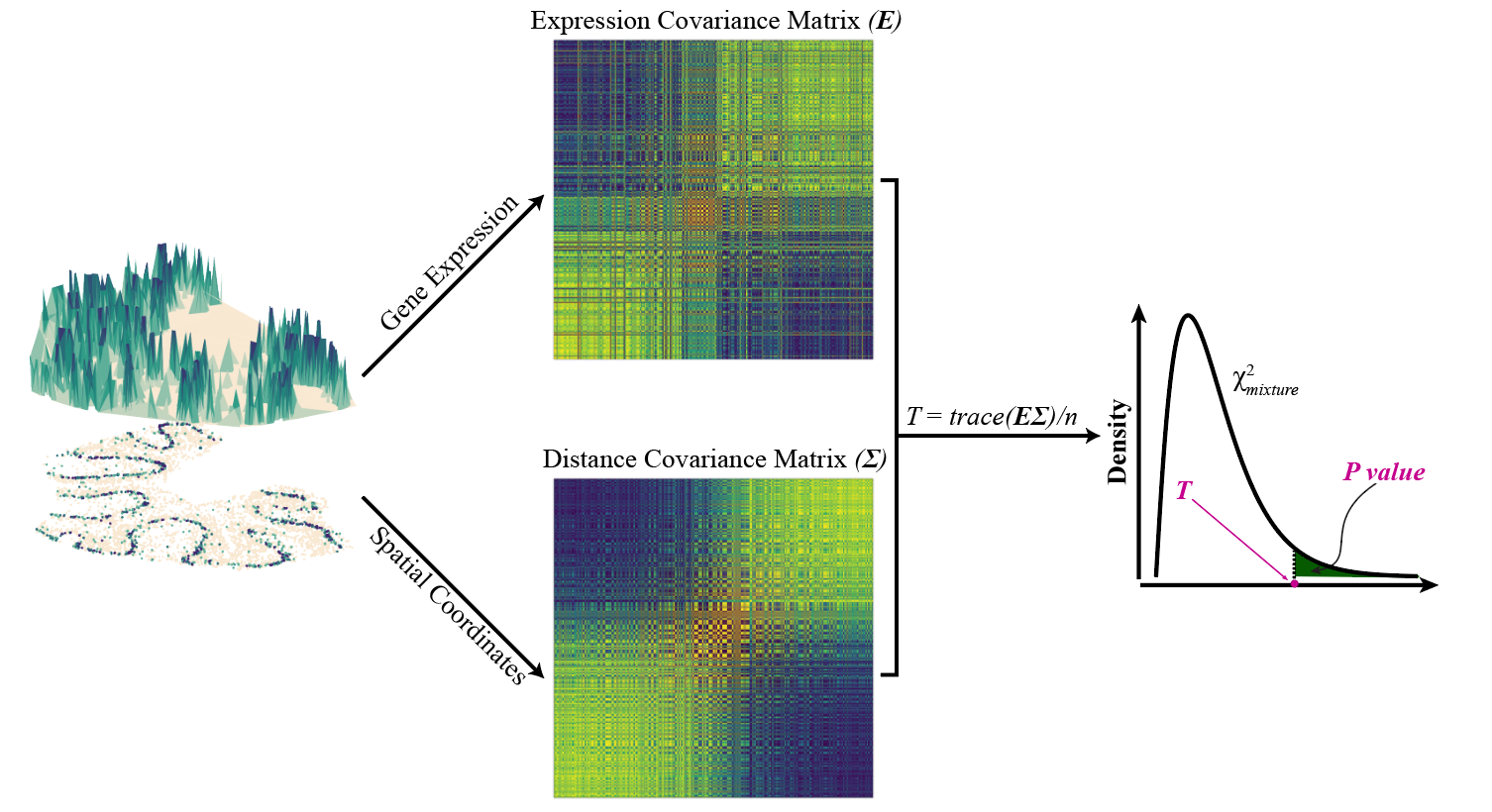 Recommend Application: Sample size greater than 3,000, works well regardless of the sparsity structure.
Recommend Application: Sample size greater than 3,000, works well regardless of the sparsity structure.
Example Analysis with SPARK-X:here.